Introduction
cfDNAPro is an R/Bioconductor package designed for the
extraction and visualization of cell-free DNA (cfDNA) features.
It provides a user-friendly framework for the automated characterization
and visualization of cfDNA sequencing data. The cfDNAPro package includes
functions for calculating overall, median, and modal fragment size
distributions, as well as identifying peaks, troughs, and the periodicity
of oscillations in the fragment size profile. Additionally, it features robust
data visualization tools.
The package can also process Copy-Number (CN) and
single point mutation information from cfDNA fragment data.
This functionality enables the integration of multiple features,
such as fragment size and mutation status, allowing for comprehensive
analysis and plotting of the data,
as demonstrated in our preprint.
The cfDNAPro package has been accepted by Bioconductor.
Motivation
Cell-free DNA (cfDNA) enters human blood circulation by various biological processes, and includes tumour-derived circulating tumour DNA (ctDNA). There is increasing evidence that differences in biological features between cfDNA and ctDNA could be exploited to improve cancer detection, treatment selection and minimal residual disease detection. However, there are currently no R packages that support analysis of cfDNA biological features such as fragment length, nucleotide frequency, nucleosome occupancy etc.
Uses and Applications
cfDNAPro can be used for a variety of cfDNA-related analysis tasks:
Cancer early detection, monitoring and therapy personalisation
Exploration of curated cfDNA biological features
Comprehensive cfDNA fragment annotation for Machine Learning Model Building
cfDNA mutation list refinement
Highlights
cfDNAPro addresses the problem regarding reproducibility
of cfDNA fragment data analysis.
The definition of “fragment length” varies across different alignment software, leading to concerns (see page 9 footnote in the SAM file format specification document SAMv1.pdf). The need for single-molecule level resolution in cell-free DNA fragmentomic analyses underscores the critical importance of precise and unbiased feature extraction.
cfDNAPro is designed to resolve this issue and standardize the cfDNA fragmentomic analysis.
As an example, we showcase how an ambiguous case occurs when there are sequence-through issues.
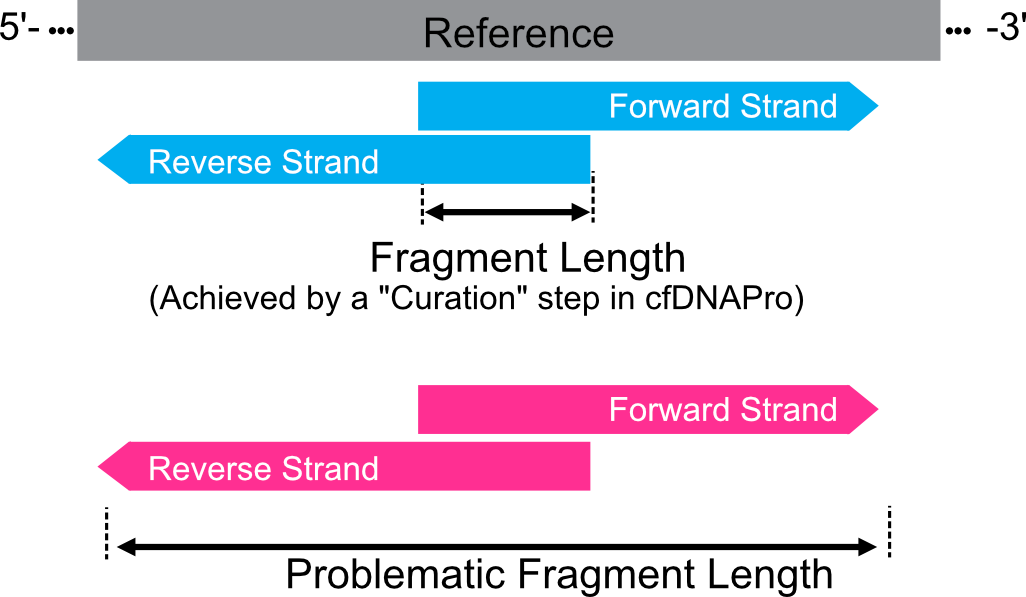
Here, we propose that the cfDNA fragment is the region between the left boundary of the forward strand and the right boundary of the reverse strand.
Input Files
Within cfDNAPro, the primary input consists of
one or more BAM files from paired-end whole-genome sequencing
(WGS) with variable depths. While the package also supports other
paired-end sequencing methods like targeted sequencing, it has not
been evaluated with these protocols.
library(cfDNAPro)
# read bam file, do alignment curation
frags <- readBam(bamfile = "/path/to/bamfile.bam")
# convert GRanges object to a dataframe in R
frag_df <- as.data.frame(frags)
Alternatively, cfDNAPro can read in insert sizes metrics files
produced by Picard Tools, using the CollectInsertSizeMetrics
tool, for fragment size analysis.
To use cfDNAPro package, gathering all txt files generated by Picard or
bam files into sub-folders named by cohort name is required,
even if when you have only one cohort. Example txt files are installed
together with this package.
Currently cfDNAPro is compatible exclusively with insert sizes metrics files produced by Picard Tools, using the CollectInsertSizeMetrics tool, which can be accessed here.
library(cfDNAPro)
path <- "path/to/main/folder"
myplot <- callMode(path = path) %>% plotMode()
If users want to access mutational fragment information, they should supply a .tsv file containing a mutation list with four columns (chr, pos, ref, alt). This will enable the annotation of each fragment’s status based on the overlap of the paired-end reads and the base of the fragment.
library(cfDNAPro)
# read bam file, do alignment curation
frags <- readBam(bamfile = "/path/to/bamfile.bam", mutation_file = "/path/to/mutations.tsv")
# convert GRanges object to a dataframe in R
frag_df <- as.data.frame(frags)
Package Usage Guide
The cfDNAPro offers a range of applications,
all of which are detailed in the tutorial section.
More details on the R function parameters can be found here.
Contact
If you have any questions about cfDNAPro, you can create an issue on github or contact haichao.wang@cruk.cam.ac.uk, paulius.mennea@cruk.cam.ac.uk.
Source code on Github
The github repository of cfDNAPro can be found at https://github.com/hw538/cfDNAPro.
Installation
Install directly in R
cfDNAPro can be installed directly in R:
if (!require(devtools)) install.packages("devtools")
library(devtools)
devtools::install_github("hw538/cfDNAPro", build_vignettes = TRUE)
Citation
If you use cfDNAPro in any published work, please cite:
Haichao Wang, Paulius D. Mennea et al (2020). cfDNAPro: An R/Bioconductor package to extract and visualise cell-free DNA biological features. R package version 1.7 https://github.com/hw538/cfDNAPro