2. Mutational Feature Processing and Visualization
2.1. Background
The terms “mutations” and “mismatches” are employed interchangeably in this context. While an alteration in matched tumor tissue may be identified as a mutation or a single-nucleotide variant (SNV), the same alteration observed in the corresponding cfDNA sample might not originate from biological processes. Instead, it could result from library preparation or sequencing artifacts. Consequently, such alterations are referred to as mismatches or single base substitutions (SBS).
In the field of cfDNA analysis, several strategies are employed to enhance the confidence that a specific base change is a tumor-derived mutation. A prevalent approach involves utilizing mutational information from matched tumor tissue as a prior. This method seeks to identify corresponding mutations in the matched cfDNA sample. If a mutation is supported by a sufficient number of unique (non-duplicate) cfDNA fragments, it is likely to be a genuine mutation present in the cfDNA sample. The probability that this mutation is tumor-derived is contingent upon the quality of the initial tumor mutation list used as a prior.
However, if the cfDNA sample displays only one fragment supporting the mutation identified in tumor tissue, this could still be an artifact arising from various stages of sample processing and sequencing. To address this, one effective denoising strategy involves the use of error-suppression techniques. An example of such a method is paired-end read overlap, which we will explore in more detail below.
If a tumor-informed approach is not feasible—meaning there is no available matched tumor tissue mutational data to use as a prior—identifying tumor-derived cfDNA mutations necessitates alternative methods. One approach requires a sufficiently high ctDNA tumor fraction, where the fraction of tumor-derived cfDNA molecules relative to all cfDNA molecules is significant enough that ≥2 cfDNA fragments support the mutation. Additional evidence must also confirm that these are not germline mutations. Alternatively, a pre-trained model can be used to differentiate ctDNA mutations from other sources of noise, such as germline variants, sequencing artifacts, and mutations due to clonal hematopoiesis. This model would leverage various ctDNA features to accurately identify tumor-specific mutations.
Currently, our package supports the analysis of SBSs. Moving forward, we aim to expand its capabilities to include additional mutation types.
cfDNAPro includes a novel feature that allows for the annotation of each
cfDNA fragment with detailed mutational information.
The package introduces an innovative functionality that allows for the annotation of each cfDNA fragment with detailed mutational information. This feature is versatile and can be employed for various applications including the identification of tumor-specific mutations in cfDNA, the refinement of mutation lists to reduce noise, and the preparation of data structures essential for training and developing deep learning/AI models.
2.2. Analyzing cfDNA Fragments with Mutational SNV Data
In this tutorial, we will demonstrate how to process BAM files to extract a range of cfDNA feature metrics alongside mutational SNV information.
The mutational data can be
provided by including an additional mutations.tsv mutation file
and specifying it in the readBam() function using the
mutation_file parameter:
# Process cfDNA data with mutational information from a pre-existing file
readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "/path/to/mutation_file.tsv")
The mutation file used in analyses can be sourced from
matched tumor tissue sequencing data or can include common cancer mutation hotspots.
It must be formatted in a tab-separated layout containing four columns:
chr pos ref alt in a tab-separated format.
Alternatively, mismatches can be identified using the wrapper function pileupMismatches(),
which utilizes the Rsamtools::pileup() function to generate a list of mismatches.
The pileup_params allows you to set the values that are forwarded to the
PileupParam() function in Rsamtools.
This method is not optimal for identifying tumour-derived mutations and may result in many false positives. Additionally, it can be inefficient in terms of computational processing and resources, as this module has not been extensively tested. Currently, it identifies the total number of cores available on your local machine and utilizes all but one of those cores to process the reads from specified chromosomes in parallel. We plan to introduce wrapper scripts for high-performance computing (HPC) systems in the near future.
The output from pileupMismatches() can be saved in a .tsv file using the pileupMismatches()
parameter outfile to indicate the path and the filename for the mismatch output.
The output mismatch list can then be used as input to the readBam() function parameter mutation_file.
# Generate mismatch list
pileupMismatches(bamfile = "path/to/bamfile.bam",
pileup_params = list(max_depth = 250,
min_base_quality = 13),
genome = BSgenome.Hsapiens.UCSC.hg38::BSgenome.Hsapiens.UCSC.hg38,
chromosome_to_keep = paste0("chr", 1:22),
galp_flag = Rsamtools::scanBamFlag(
isPaired = TRUE,
isDuplicate = FALSE,
isSecondaryAlignment = FALSE,
isUnmappedQuery = FALSE,
isSupplementaryAlignment = FALSE),
num_cores = 3,
outfile = "path/to/mismatch_file.tsv")
# Process cfDNA data with mutational data from Rsamtools pileup generated list
readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "path/to/mismatch_file.tsv",
mut_fragments_only = FALSE,
genome_label = "hg38",
chromosome_to_keep = paste0("chr", 1:22),
galp_flag = Rsamtools::scanBamFlag(
isPaired = TRUE,
isDuplicate = FALSE,
isSecondaryAlignment = FALSE,
isUnmappedQuery = FALSE,
isSupplementaryAlignment = FALSE))
An alternative approach would be to generate a mismatch list
using bcftools mpileup in bash and then provide
it as a .tsv file for the readBam() function.
The default behavior of the readBam() function is to process all DNA fragments
within the BAM file. However, by setting mut_fragments_only = TRUE,
the function will only analyse fragments that overlap with the specified mutation loci.
This option reduces the computational load and may be adequate for users who do not
require information from non-mutation overlapping fragments/read pairs.
2.3. Advantages of the readBam() Function
The readBam() function will read, extract,
and process cfDNA fragment data,
combining it with the mismatch or SNV (i.e., mutational information) data.
Each fragment will be annotated if it overlaps a mutation locus,
indicating whether the fragment’s base matches the REF or ALT base.
An additional advantage of this mutational feature is that paired-end overlap information is also considered. cfDNA fragments vary in size, and in a typical 150bp paired-end sequencing run, the paired reads will have different lengths of overlap. The scenarios of overlap are illustrated below:
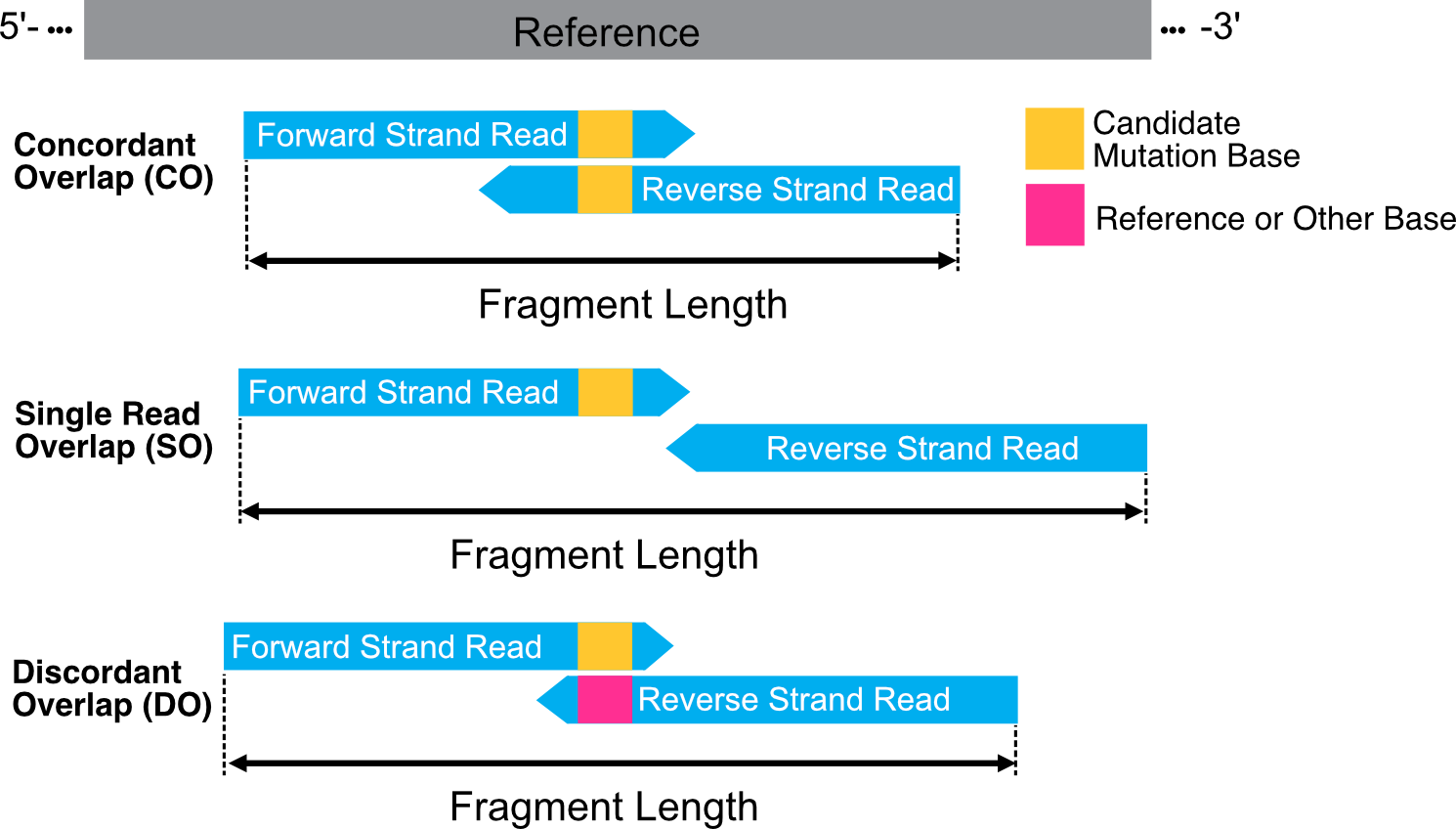
Sequencing errors occurring during the nucleotide addition stage in the sequencing flowcell can be suppressed by selecting mismatches/mutations that are supported by both paired-end reads.
Therefore, the readBam() function not only
annotates each fragment/read-pair with information on
whether it covers a mutation locus and whether it has
the REF or MUT base, but also includes paired-end
read overlap information.
This indicates whether only one of the paired-end reads covers the mutation
locus (SO - single-read overlap), or both reads overlap the mutation locus
with concordant mutation locus bases (CO - concordant read overlap) or
discordant bases (DO - discordant read overlap).
Below are the different scenarios for running the readBam() function:
# Process all fragments present within the BAM file without mutational annotation
readBam(bamfile = "path/to/bamfile.bam",
mutation_file = NULL)
# Process all fragments present within the BAM file with additional mutation annotation
readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "/path/to/mutation_file.tsv",
mut_fragments_only = FALSE)
# Process fragments that overlap loci indicated in the mutation file
readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "/path/to/mutation_file.tsv",
mut_fragments_only = TRUE)
2.4. Mutation Output Files
The mismatch/mutational information will
be encoded within the GRanges object
along with other columns indicating
fragment ID, fragment length,
mutation status of whether it overlaps a target mutation locus,
the paired-end read overlap type, and the base type.
The GRanges object can then be converted to a dataframe
for further inspection based on your analysis.
# Process the BAM File
frag_obj <- readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "/path/to/mutation_file.tsv")
# Convert GRanges object to dataframe for further inspection
frag_obj <- as.data.frame(frag_obj)
Additionally, each mutationally annotated GRanges object
can be exported as a mutation-oriented .tsv file:
# Process BAM File
frag_obj <- readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "/path/to/mutation_file.tsv")
# Export a mutation-oriented .tsv table summarising fragment information per mutation locus
writeMutTable(granges_object = frag_obj, output_file = "./table.tsv")
The mutation-oriented table will summarize the number of fragments for each candidate mutation, categorizing the fragments by their base type and read-pair overlap type.
Lastly, we can visualise the the 96 trinucleotide SBS profile of each sample,
by utilising the callTrinucleotide and plotTrinucleotide functions.
For each mutation locus, we will summarize the mutational information by
identifying the most prominent mutant base if multiple different bases are
present and by selecting the most prominent read overlap type.
If the mutation is supported by a discordant overlap fragment,
then we will use the base that supports the base from the mismatch/mutation list.
The output_file and ggsave_params parameters can be used to save the output
with the desired specifications:
output_file: String; the name and path of the output PDF file.ggsave_params: A list of parameters to be passed toggplot2::ggsave(). This list can include any of the arguments thatggsave()accepts. Default settings ofggsave()will be used unless specified. Example:list(width = 10, height = 8, dpi = 300)
# Process cfDNA and Mutational Data
frag_obj <- readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "/path/to/mutation_file.tsv")
# Generate a Dataframe with Trinucleotide SBS Information
trinuc_obj <- callTrinucleotide(frag_obj)
# Plot the Trinucleotide SBS Profile
plotTrinucleotide(trinuc_obj,
output_file = "./trinucleotide_profile.pdf",
ggsave_params = list(
width = 17,
height = 6,
units = "cm",
device = "pdf"))

The plot will discriminate between SBS supported by discordant-bases, and single-read/paired-read overlap.
Alternatively, we can plot the trinucleotide profile by excluding the read-pairs with discordant bases. A mutation locus, such as chr22:50592320-50592320:A-G, can have a different number of read-pair types covering it. The types are:
CO_MUT: Concordant read-pair overlap with mutation baseSO_MUT: Single read overlap with mutation baseCO_REF: Concordant read-pair overlap with reference baseSO_REF: Single read overlap with reference baseDO: Discordant read-pair bases at mutation locusSO_OTHER: Single read overlapping a base that is neither the REF nor MUT base at the indicated mutation locusCO_OTHER: Concordant read-pair overlap at mutation locus but supporting a base that is neither REF nor MUT
By using the plotTrinucleotide() function’s remove_type parameter, we can
indicate that for each SBS locus, we want to set all discordant read-pair overlaps
(DO) to 0.
# Process cfDNA and Mutational Data
frag_obj <- readBam(bamfile = "path/to/bamfile.bam",
mutation_file = "/path/to/mutation_file.tsv")
# Generate a Dataframe with Trinucleotide SBS Information
trinuc_obj <- callTrinucleotide(frag_obj)
# Plot Trinucleotide SBS Profile by removing the discordant read-pair overlaps
plotTrinucleotide(trinuc_obj,
remove_type = c("DO"),
output_file = "./trinucleotide_profile.pdf",
ggsave_params = list(
width = 17,
height = 6,
units = "cm",
device = "pdf"))

If we want to remove every locus that had even one discordant
read-pair supporting that mutation, we can use the
exclude_if_type_present = c("DO") parameter.
Conversely, if we want to retain every SBS that had even one concordant
read-pair overlap supporting it, we can use the
retain_if_type_present = c("CO") parameter.
# Plot Trinucleotide SBS Profile by excluding loci with discordant read-pair overlaps
plotTrinucleotide(trinuc_obj,
exclude_if_type_present = c("DO"))
# Plot Trinucleotide SBS Profile by retaining loci with concordant read-pair overlaps
plotTrinucleotide(trinuc_obj,
retain_if_type_present = c("CO"))